
Prof. Lev Kantorovich from the School of Natural and Mathematical Sciences, King’ s College London will give a seminar on Wednesday the 13th of April, at 2 pm (room ENG209 in the Engineering Hub, building 13 on the university map). Please join us in attending this interesting talk on graphene growth.
Theoretical modelling of early stages of graphene growth by epitaxial methods
One method of growing epitaxial graphene is temperature programmed growth (TPG). In this method hydrocarbon molecules are deposited onto a transition metal surface at room temperature and then the temperature is increased in order to facilitate the thermal decomposition of the hydrocarbons and lead to the formation of graphene flakes.
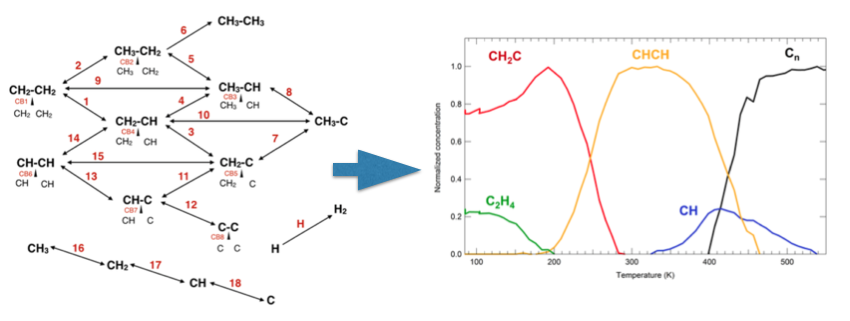
The thermal decomposition mechanism of ethylene was investigated with a combined approach of experimental and theoretical techniques. X-ray photoelectron spectroscopy (XPS) experiments were used along with core level binding energy calculations to identify the evolution of species on the Ir(111) surface as the temperature is increased. A complete reaction scheme incorporating all possible reactions between the various CnHm species, from ethylene to C monomers and dimers, was also developed. The energy barriers for each reaction were calculated using the DFT based nudged elastic band method. These were then used to simulate the kinetics and determine the species evolution on the surface. This resulting temperature evolution agrees with the photoemission measurements. The molecular dissociation mechanism begins with the dehydrogenation of ethylene to vinylidene (CH2C), which is then converted to acetylene (CHCH) by the removal and addition of an H atom. The C-C bond is then broken to form methylidyne (CH), which finally dehydrogenates to produce C monomers that are available for the early stage nucleation of the graphene islands.
Following from this nucleation of carbon clusters prior to the formation of graphene islands was also investigated. The number of carbon atoms in the critical cluster, which is equally likely to grow or to shrink in the prevailing conditions, was determined by the considering appropriate nucleation free energy. Using ab initio density functional theory calculations, the free energies of carbon clusters (containing up 16 C atoms) on the Ir(111) surface were calculated based on the configurational and vibrational contributions. The results are strongly dependent on temperature, showing its importance to cluster growth. Furthermore, we find that different types of cluster (linear, compact, dome etc.) are more stable over different size ranges. Based on this mechanisms which allow the clusters to reconstruct their structure are investigated.

Reblogged this on Maths & Physics News.